
Bassa statura: che cos’è e come si manifesta
Una crescita normale riflette uno stato generale di benessere del bambino. Una crescita ridotta può invece far sospettare un problema alla base. Quindi, una bassa statura impone sempre un attento esame al fine di riconoscere precocemente quei pazienti con patologie specifiche per le quali potrebbe essere indicata una terapia sostitutiva. Una bassa statura può essere riscontrata a qualsiasi età, ma più frequentemente in età scolare, epoca in cui è più evidente il confronto con i coetanei. A volte, però, può accadere che il problema della statura si evidenzi successivamente quando, cioè, la statura del bambino, fino ad allora normale, diventa inferiore rispetto a quella dei coetanei. Un rallentamento del ritmo accrescitivo dal percentile originario può essere la prima manifestazione di patologia cronica oppure endocrinologica e deve essere tempestivamente indagato da parte del Pediatra curante. Tra le molteplici cause di bassa statura le principali sono:
- La bassa statura idiopatica
- La bassa statura famigliare
- Il ritardo di crescita intrauterina
- Il ritardo costituzionale di crescita e pubertà
- Le sindromi genetiche malformative
- Le cromosomopatie
- L’aploinsufficienza del gene SHOX
- Le malattie metaboliche
- Le malattie endocrine
- Le malattie croniche
- Le malattie ossee
Cosa fare?
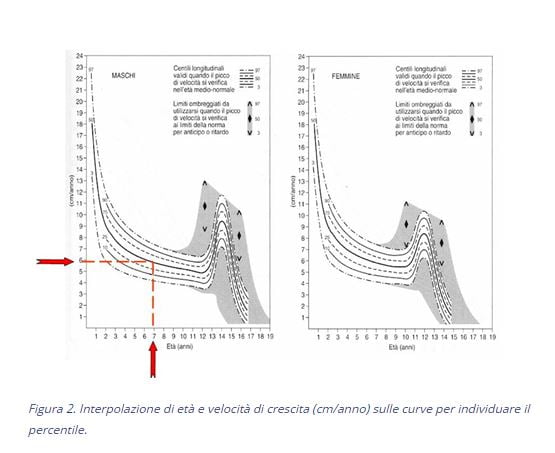
Di fronte al bambino che cresce stentatamente e che é sempre “il più piccolo della classe”, i genitori si rivolgono in prima istanza al Pediatra di famiglia, il quale valuta accuratamente la statura del bambino per rispondere alla pressante richiesta dei genitori di “fare qualcosa per farlo crescere”. Utilizza a questo scopo strumenti adeguati quali stadiometri ed infantometri (questi ultimi per i bambini di età inferiore ai 2 anni che vengono misurati supini non deformabili con il tempo. Intersecando l’altezza del bambino con la sua età cronologica su apposite grafiche differenziate per maschi e per femmine, che riportano la crescita dalla nascita all’età adulta di una popolazione normale della stessa etnia, il pediatra individua il percentile corrispondente, evidenziando eventuali deviazioni dalla normalità. (Figura 1 e 2)

Alla rilevazione della statura e della velocità di crescita, si deve aggiungere la valutazione del “target genetico”, utilizzando la formula:
[(altezza padre+altezza madre)/2] +6.5 cm (se maschio) oppure –6.5 cm (se femmina)
Questo parametro auxologico è utile per selezionare i bambini con bassa statura familiare. Inoltre, è necessaria la valutazione dello sviluppo puberale. Quest’ultimo viene schematizzato in cinque stadi nei confronti di:
- peluria pubica (pubic hair, PH),
- sviluppo mammario (breast, B)
- morfologia dei genitali maschili (genitalia, G). Il volume testicolare viene quantificato utilizzando l’orchidometro di Prader e inserito sugli appositi percentili per giudicarne la normalità in base all’età.
Nei casi di pubertà ormai avanzata (comparsa del menarca nella femmina e volume testicolare già corrispondente a quello osservato in età adulta nel maschio), gli accertamenti risultano inutili poiché la spinta accrescitiva staturale è già terminata, come può essere confermata radiologicamente dalla saldatura delle cartilagini di accrescimento.
Bambini con statura estremamente bassa
Se il bambino presenta una statura significativamente bassa (esempio, inferiore al 3° percentile con target genetico normale) oppure se sono già presenti i primi segni puberali (comparsa della ghiandola mammaria nella femmina ed aumento del volume testicolare nel maschio), si inizia subito l’iter diagnostico, senza aspettare il monitoraggio della velocità di crescita. Anche nel caso in cui il bambino presenti una statura al di sotto del percentile corrispondente al target genetico, cioè sia “troppo piccolo in confronto ai genitori”, si può iniziare l’iter diagnostico subito oppure si può monitorare la sua velocità di crescita ogni sei mesi. Se il bambino, ancora in epoca prepuberale, presenta una statura ai limiti inferiori della norma ma cresce lungo lo stesso percentile, cioè mantiene la stessa velocità di crescita, non è necessario iniziare subito l’iter diagnostico. In questo caso è sufficiente monitorare il suo ritmo di crescita con controlli auxologici semestrali e la progressione dello sviluppo puberale.
In caso contrario, cioè quando il ritmo di crescita si allontana dal “binario percentilico” fino ad allora seguito, si inizia l’iter diagnostico.
Di fronte ad un bambino con bassa statura, bisogna stabilire se questa sia armonica o disarmonica. Nel soggetto normale, in posizione eretta, le estremità delle dita raggiungono la metà o i due terzi inferiori della coscia. Un soggetto è viceversa disarmonico se le estremità delle dita si trovano in corrispondenza della radice della coscia oppure raggiungono le ginocchia.
Diagnosi
Una volta accertata la bassa statura, l’iter diagnostico continua con la raccolta di un’accurata anamnesi familiare, finalizzata alla ricerca di condizioni patologiche ereditarie che possono determinare una bassa statura nel bambino. Occorre, pertanto, indagare l’eventuale presenza di altri casi di bassa statura nell’ambito della famiglia. La maggioranza dei bambini con statura pari oppure inferiore al 3° centile appartengono a due condizioni che possono essere considerate delle varianti normali del processo di crescita: la bassa statura familiare e il ritardo costituzionale di crescita. La raccolta di una completa anamnesi familiare e personale aiuta ad inquadrare questi soggetti. Infatti, un bambino con bassa statura familiare ha generalmente entrambi i genitori o almeno uno dei due con un’altezza inferiore alla norma. Nei soggetti con ritardo costituzionale di crescita, la pubertà inizia più tardi rispetto ai coetanei: oltre i 13 anni nelle femmine e i 14-15 anni nei maschi. Una volta iniziata, la pubertà procede regolarmente e questi adolescenti raggiungono la statura finale più tardi rispetto ai coetanei. In anamnesi familiare spesso emerge che uno o entrambi i genitori hanno presentato un ritardo puberale.
In alcuni casi, peraltro molto rari, può essere presente una storia familiare di deficit di GH, quando i genitori sono particolarmente piccoli e non sono stati precedentemente indagati dal punto di vista endocrinologico. Infine, l’anamnesi patologica remota deve indirizzare verso condizioni croniche (esempio, nefropatie, emopatie, malattie infiammatorie intestinali croniche, ecc).
Un attento esame obiettivo del paziente deve essere mirato all’osservazione dell’aspetto generale del bambino e al suo stato di nutrizione, che deve essere indagato calcolando il Body Mass Index il quale si ottiene dividendo il peso corporeo (espresso in Kg) per l’altezza (espressa in metri) elevata al quadrato e deve essere quantificato utilizzando specifici grafici di normalità per l’età pediatrica.
Vanno ricercate la presenza di eventuali malformazioni corrette chirurgicamente (ad esempio, una cheilo-palatoschisi) e il periodo di insorgenza del rallentamento/arresto della curva di crescita. Ad esempio, se l’arresto della crescita si è verificato improvvisamente, a volte accompagnato da sintomi d‘ipertensione endocranica (esempio, una cefalea, un vomito a digiuno, disturbi visivi), si deve considerare la possibilità di un processo espansivo endocranico (ad esempio un craniofaringioma, medulloblastoma, ecc) da indagare con esami neuroradiologici. Un attento esame obiettivo deve poi permettere di riscontrare segni o sintomi caratteristici di alcune condizioni peculiari o sindromi legate ad anomalie cromosomiche, quali la sindrome di Turner, la sindrome di Down oppure sindromi non cromosomiche, quali la sindrome feto alcolica.
Altre informazioni riguardano l’eventuale presenza di patologie legate all’ambiente socio-economico, quali parassitosi intestinali facilitate da condizioni di vita disagiate oppure malnutrizione, che potrebbero essere alla base di una scarsa crescita del bambino. A tal proposito, è stato dimostrato che i bambini più poveri tendono ad essere più piccoli rispetto a quelli appartenenti alle classi socio-economiche più elevate. Inoltre, anche la deprivazione emotiva estrema può causare un ritardo di crescita, determinando un’inibizione ipotalamico-ipofisiaria reversibile. In questi casi, l’ambiente familiare è alterato e il bambino appare vittima di abusi e trascuratezza. La sua crescita riprende rapidamente dopo che viene allontanato dall’ambiente oppressivo.
Parametri auxologici alla nascita
Infine, vanno considerati i parametri auxologici alla nascita. I soggetti che durante la vita fetale presentano uno scarso accrescimento costituiscono un gruppo molto eterogeneo (nati piccoli per l’età gestazionale, SGA) sia per quanto riguarda l’eziopatogenesi del loro ritardo staturale, sia per il decorso della loro crescita. La maggioranza recupera il deficit di accrescimento entro 6-24 mesi dalla nascita, mentre altri soggetti mantengono il deficit iniziale o addirittura lo peggiorano durante la vita extrauterina.
Una volta accertata una bassa statura non riconducibile a condizioni clinicamente evidenziabili, quali ad esempio un’acondroplasia, si procede ad uno screening di primo livello volto ad escludere le principali e più diffuse patologie endocrino-metaboliche quali la celiachia, il malassorbimento e l’ipotiroidismo subclinico. Devono, poi, essere escluse cause sistemiche di bassa statura che, se non trattate o scarsamente controllate, possono avere un effetto negativo sulla crescita, quali nefropatie, epatopatie ed altre patologie d’organo. In età pediatrica, ad esempio, l’insufficienza renale cronica, comporta spesso gravi disturbi dell’accrescimento. Secondo il grado di compromissione della funzione renale l’iposomia può essere più o meno marcata ed accompagnata ad altri disturbi del metabolismo osseo. Nonostante le migliorate possibilità terapeutiche, quali la dialisi o il trapianto di rene non sempre viene ristabilita una corretta ripresa dei processi di accrescimento. Dopo i risultati dei primi studi sulla somministrazione di GH nei bambini con insufficienza renale cronica, oggi tale patologia fa parte di quelle condizioni cliniche in cui l’uso della terapia con ormone della crescita è regolarmente previsto dalle nostre norme sanitarie (Nota AIFA 39).
Nel paziente affetto da beta-talassemia, la crescita staturo-ponderale, generalmente normale fino ai nove anni di età, decelera successivamente, con rischio di una statura finale più bassa rispetto al target genetico. Una statura inferiore al 3° centile è presente in più del 30% dei pazienti talassemici che hanno raggiunto l’età adolescenziale. Le cause non sono state del tutto chiarite, ma vari meccanismi eziopatogenici sono stati considerati, tra cui anemia cronica, ipersplenismo, disordini endocrini secondari al sovraccarico marziale (insufficienza/deficienza di GH, ipotiroidismo, ipogonadismo), epatopatia cronica, displasia scheletrica secondaria a “tossicità di Desferrioxamina”.
Nella diagnosi differenziale tra le condizioni di bassa statura è utile la valutazione dell’età ossea che è un importante indice di crescita in età pediatrica. Nel bambino normale l’età ossea può coincidere con la cronologica oppure essere di poco ritardata. Rappresenta un parametro predittivo della statura che il soggetto raggiungerà in età adulta. L’età ossea viene valutata sulla radiografia della mano sinistra (o del ginocchio e del piede nei primi mesi di vita), confrontandola con le immagini-standard dell’Atlante di Greulich e Pyle. Questo, che è il metodo più utilizzato per la rapidità della valutazione, tiene conto del numero e delle dimensioni delle ossa della mano e del polso. L’età ossea può essere quantificata anche utilizzando un metodo più indaginoso, quello di Tanner e Whitehouse in cui si valutano le singole ossa dando un punteggio a ciascuna secondo lo stadio di maturazione e riportando le misurazioni su una carta dei percentili. A tal proposito è importante sottolineare che il calcolo dell’età ossea è dipendente dall’esperienza dell’operatore. Generalmente, nel deficit di GH, la maturazione ossea è ritardata in accordo con la severità e la durata del deficit stesso. L’esecuzione di un cariogramma sarà indicato nel caso di una bambina la cui bassa statura non sia inquadrabile in una condizione nota al fine di escludere una sindrome di Turner oppure un mosaicismo turneriano, e nel caso di un maschio che presenti dismorfie somatiche associate a bassa statura.
Una volta escluse le cause non endocrine di bassa statura, in presenza di parametri auxologici suggestivi per un deficit di GH, appare indicato inviare il soggetto ad un Centro specialistico di Auxo-endocrinologia Pediatrica, per ulteriori accertamenti mirati a studiare l’asse ipotalamo-ipofisi-IGF-I. Si valuterà la secrezione di GH dopo stimolo farmacologico, il livello di IGF-I circolante e la risonanza magnetica nucleare encefalica per evidenziare eventuali alterazioni della regione ipotalamo-ipofisaria. Il deficit di GH può essere isolato oppure associato ad altre tropine ipofisarie.
L’ ipopituitarismo congenito è caratterizzato da deficit multiplo degli ormoni ipofisari, come il deficit dell’ormone somatotropo, tireotropo, lattotropo, corticotropo o gonadotropo, causato da mutazioni dei fattori di trascrizione coinvolti nell’ontogenesi dell’ipofisi. La conferma della diagnosi avviene tramite sequenziamento diretto dei geni dei fattori di trascrizione. L’ipopituitarismo congenito è causato da mutazioni di diversi geni che codificano per i fattori di trascrizione. Il fenotipo varia a seconda del fattore di trascrizione coinvolto: PROP1 (deficit degli ormoni somatolattotropo tireotropo, gonadotropo e a volte corticotropo; iperplasia e ipoplasia ipofisaria), POU1F1 (carenza degli ormoni somatolattotropo e tireotropo, ipoplasia ipofisaria), HESX1 (carenze ipofisarie variabili, displasia setto-ottica), e meno frequentemente LHX3 (deficit degli ormoni somatolattotropo e gonadotropo, limitazioni nella rotazione del capo e del collo) e LHX4 (deficit ipofisari variabili, ectopia della neuroipofisi, anomalie dell’encefalo). Nel trattamento sostitutivo é necessaria un’appropriata integrazione delle carenze ormonali. Recentemente sono state messe a punto indagini genetiche, note come micro array CGH, che possono evidenziare delezioni e/o duplicazioni dei geni legate a bassa statura.
L’iter diagnostico è in continua evoluzione grazie al miglioramento delle tecniche di indagine genetica.
Le domande dei genitori e come rispondere
- A nostra figlia è comparso il ciclo mestruale nella femmina. Vuol dire fine di crescita? No, significa solo fine dell’accelerazione puberale e in genere vi può essere ancora un guadagno in altezza di circa 5 centimetri.
- Posso calcolare quanto crescerà mio figlio? La crescita staturale dipende da molti fattori, tra cui l’altezza dei genitori. Non è possibile predire con certezza quanto diventerà alto un bambino/a, ma si può calcolare quanto potrà ancora crescere in base alla progressione della maturazione scheletrica. È necessario tenere presente che sono calcoli su base statistica, quindi teorici. Si può calcolare però il suo “target genetico”, utilizzando la formula [(altezza padre+altezza madre)/2] +6.5 cm (se maschio) oppure –6.5 cm (se femmina). Il valore corrisponde alla statura che i genitori hanno programmato geneticamente. Quindi sarà uguale per tutti i maschi e tutte le femmine nate dalla stessa copia. Quindi calcoli teorici.
- Si può prescrive la terapia con GH dopo la pubertà in un bambino di bassa statura per farlo crescere in altezza? Terminata la pubertà, con la chiusura delle cartilagini di accrescimento osseo, non è possibile utilizzare il GH per aumentare la statura definitiva. In ogni caso, la sua prescrizione è regolamentata da severe norme nazionali.
- Che cosa è il GH che viene usato a scopo terapeutico? Il GH è una proteina che in natura prodotta dalle cellule dell’ipofisi. Però oggi é prodotto solo con tecniche di bioingegneria, e più specificamente con il metodo del “DNA ricombinante”. Esso ha una struttura uguale a quella umana e un livello di sicurezza terapeutica molto elevato.